A recent study tried to figure out what this chromosome actually does…
Yesterday, you could read about the crazy sex-chromosomes of larks. It turned out that the sex-chromosomes of these birds consists of parts from several other chromosomes. Weird… A recent study in the journal Nature Communications revealed another chromosomal conundrum in songbirds: the so-called germline restricted chromosome (or GRC for friends). Let’s get acquainted.

What mysteries does the GRC of this Zebra Finch (Taeniopygia guttata) hold? © Peripitus | Wikimedia Commons
An Accessory Chromosome
The story of the GRC starts with two researchers investigating the karyotype of the Zebra Finch (Taeniopygia guttata). In the germline (i.e. cells in the testes and ovaries) of this bird, they found a big “accessory” chromosome. Surprisingly, in the rest of the body cells – the so-called soma – this chromosome was absent. Further investigations revealed that, in males, it is eliminated during the formation of sperm cells. In females, however, the chromosome is passed on to the offspring but gets eliminated in soma-cells. Good riddance, chromosome! Because this extra chromosome only resides in the germline, it was dubbed germline-restricted chromosome (GRC).

The karyotypes seven biggest chromosomes and sex chromosomes (ZW) of Zebra Finch for (a) female soma, (b) male some, and (c) male germline. The big germline-restricted chromosome (GRC) is highlighted with the arrow. From: Pigozzi & Solari (1998) Chromosome Research
Which Genes?
The presence of the GRC raises a lot of questions. First of all, what kind of genes are on this chromosome? The researchers of the recent study predicted that the GRC would be enriched in repetitive elements, a kind of wastebasket for harmful “genetic parasites”. Surprisingly, the GRC contained numerous genes from other chromosomes (i.e. paralogs). Using a clever combination of the latest genome-sequencing techniques and some bioinformatic wizardry, they succeeded in pinpointing 115 high-confidence GRC-linked genes. These genes were copied to the GRC from 19 chromosomes (18 autosomes and the Z-chromosome).
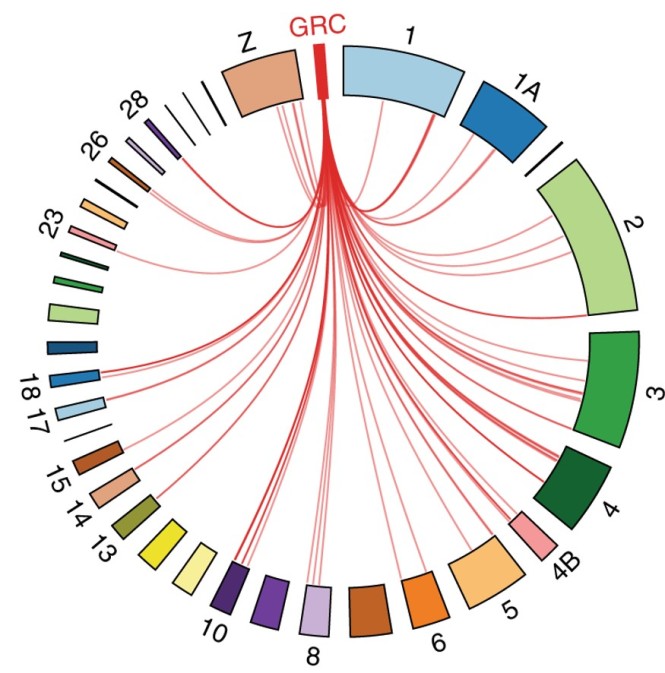
The GRC contains gene from numerous other chromosomes. From: Kinsella, Ruiz-Ruano et al. (2019) Nature Communications
Expressed Genes?
The presence of these genes does not necessarily mean that they are actively used. Perhaps the GRC is a reservoir for decaying pseudogenes? To infer whether these genes are active, the researchers turned to transcriptomic (RNA) and proteomic (proteins) data. These analyses revealed that at least 6 genes were expressed in testes and 32 in ovaries. Because some GRC-genes are (almost) identical to their paralogs on other chromosomes, the researchers could not distinguish between them. Thus, there might be more active GRC-genes.
The divergence between some GRC-genes and their paralogs already indicates that different genes were copied to the GRC at different times. By comparing the DNA-sequence of GRC-genes and paralogs, we can deduce how the GRC changed over time. Some genes, such as bicc1 and trim71, were early additions during the diversification of songsbirds (more than 25 million years ago). Other genes were recently added to the mix. The figure below gives a nice overview of the gradual accumulation of genes on the GRC.

The GRC captured different genes during its evolutionary history. From: Kinsella, Ruiz-Ruano et al. (2019) Nature Communications
What does it do?
The GRC is not restricted to the Zebra Finch. A recent study in PNAS found this chromosome in 15 other songbirds, suggesting that it plays an important role in the germline of these species. The presence of several germline developmental genes indicates that it might act as a germline-determining chromosome. The authors also speculate that “a GRC may allow adaptation to germline-specific functions free of detrimental effects on soma.” In other words, the GRC could prevent conflicts between germline and soma.
Finally, the observation of several species-specific genes on the GRC supports the idea that it might contribute to reproductive isolation during the formation of new species. Could the GRC explain the massive diversification of songbirds?
References
Kinsella, C.M., Ruiz-Ruano, F.J., et al. (2019) Programmed DNA elimination of germline development genes in songbirds. Nature Communications, 10:5468.
Pigozzi, M. I., & Solari, A. J. (1998). Germ cell restriction and regular transmission of an accessory chromosome that mimics a sex body in the zebra finch, Taeniopygia guttata. Chromosome Research, 6(2):105-113.
Torgasheva, A. A. et al. (2019). Germline-restricted chromosome (GRC) is widespread among songbirds. Proceedings of the National Academy of Sciences, 116(24), 11845-11850.